





EVERY
WOMAN HAS THE
WOMAN HAS THE

TO CHOOSE HER PATH FORWARD
Actual breast reconstruction patient with Natrelle INSPIRA® Breast Implants.
Individual results may vary.

Supporting
women and their breast
cancer journeys
If you’ve decided to have reconstruction with breast implants,
Natrelle® has a wide range of innovative offerings designed to meet your expectations.
LISA
“Reconstruction after breast cancer is a journey.
It helped me discover my new normal.
It was my choice and
IT’S EXACTLY WHAT I WANTED.”
Actual breast cancer and breast
reconstruction patient with
Natrelle INSPIRA® Breast Implants.
Individual results may vary.
reconstruction patient with
Natrelle INSPIRA® Breast Implants.
Individual results may vary.
KAYLA
“After testing positive for BRCA2, I decided to have a double mastectomy and reconstruction with implants. Breast reconstruction helped me
MOVE FORWARD.”
Actual breast reconstruction patient with
Natrelle INSPIRA® Breast Implants.
Individual results may vary.
Natrelle INSPIRA® Breast Implants.
Individual results may vary.
Your GUIDE to surgery
Breast reconstruction basics
When you have a mastectomy, you lose breast tissue, some skin, and possibly your nipple and areola.
Breast reconstruction results have improved over the years. While your plastic surgeon can’t give you the same breast
you had prior to your mastectomy, he or she may be able to give you a reasonable likeness of your premastectomy breast.
Immediate or delayed reconstructive surgery
Will you begin your reconstruction surgery immediately—at the time of your mastectomy—or will you delay your reconstruction surgery until after you have healed?
Advantages
Disadvantages
Your surgeon will determine if you are an appropriate candidate for immediate reconstruction or
if it would be better for you to have a delayed procedure.
Actual breast cancer and breast reconstruction patient with Natrelle INSPIRA® Breast Implants.
Individual results may vary.
I’M
TRUSTING MY
journey
Implant-based breast reconstruction
This type of reconstruction shapes the breast with implants and is usually performed in
2 stages: Using a tissue expander in the first stage and then inserting a breast implant under the muscle in the second stage.
An innovative advancement allows for placement of a tissue expander or silicone breast implant over the chest muscle (prepectorally). Learn more about prepectoral reconstruction below, and talk to your surgeon to determine whether this approach is an option for you.
An innovative advancement allows for placement of a tissue expander or silicone breast implant over the chest muscle (prepectorally). Learn more about prepectoral reconstruction below, and talk to your surgeon to determine whether this approach is an option for you.
Advantages of Implant-based Reconstruction
Disadvantages of Implant-based Reconstruction
i am

what i choose
Actual breast cancer and breast reconstruction patient with Natrelle INSPIRA® Breast Implants.
Individual results may vary.
Over the muscle (pre-pec) technique
This technique allows for placement of a tissue expander or silicone-filled breast implant over the chest muscle (prepectorally).
Advantages of
Pre-pec Technique
Disadvantages of
Pre-pec Technique
Subpectoral (under the muscle) technique
This technique involves placing a breast implant under the chest muscle. In some cases, this can be done immediately after
the mastectomy.
If you have minimal breast tissue, your surgeon may recommend placing a tissue expander under your skin and chest muscle before placing the implant. A tissue expander is like a balloon, which can be inflated to stretch the skin and muscle over several months, making room for your implant.
If you have minimal breast tissue, your surgeon may recommend placing a tissue expander under your skin and chest muscle before placing the implant. A tissue expander is like a balloon, which can be inflated to stretch the skin and muscle over several months, making room for your implant.
Before
Primary reconstruction with sub-pec placement and animation deformity when flexing the chest muscle
After
Revision reconstruction to pre-pec placement to correct animation deformity
Before
Primary reconstruction with sub-pec placement and animation deformity when flexing the chest muscle
After
Revision reconstruction to pre-pec placement to correct animation deformity
Before
Primary reconstruction with sub-pec placement and animation deformity when flexing the chest muscle
After
Revision reconstruction to pre-pec placement to correct animation deformity
Before
Primary reconstruction with sub-pec placement and animation deformity when flexing the chest muscle
After
Revision reconstruction to pre-pec placement to correct animation deformity
Advantages of
Sub-pec Technique
Disadvantages of
Sub-pec Technique
Two-stage vs one-stage breast reconstruction
Two-stage breast reconstruction is the most common type of breast reconstruction performed. The first stage involves placing a tissue expander to recreate a breast pocket after the breast tissue has been removed. In the second stage, the implant is placed into the breast pocket.
One-stage, or direct-to-implant (DTI), breast reconstruction involves placing the implant immediately after breast tissue has been removed.
One-stage, or direct-to-implant (DTI), breast reconstruction involves placing the implant immediately after breast tissue has been removed.
Advantages of
DTI Reconstruction
DTI Reconstruction
Disadvantages of
DTI Reconstruction
DTI Reconstruction
First stage
This procedure first uses a tissue expander, which is surgically inserted and slowly expanded over time to create a space for the implant. Typically, you will meet with your plastic surgeon every other week to fill the expander with sterile saline in order to gradually stretch the skin.

The tissue expander with
over 30 years of clinical experience
Natrelle® 133S Smooth Tissue Expanders are made to match
Natrelle INSPIRA® Round Gel Breast Implants. This means that the tissue expander creates a space customized for the breast implant.
Second stage
Once the space reaches the correct size and the tissue has healed, a second surgery will remove the tissue expander and replace it with the
breast implant. If a mastectomy was only performed on one breast, some patients choose to have the other breast augmented to maintain symmetry.

The #1 surgeon-selected gummy breast implant collection in the US*
Natrelle INSPIRA® Round Gel Breast Implants
are designed to give you the look and feel that’s important to you.
*Based on surgeon survey data, September 2021 (N = 324).
Your choice. Your look.
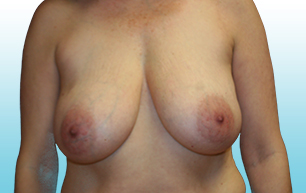
before
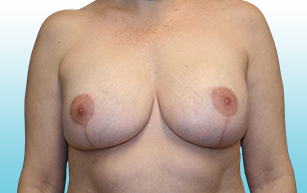
After
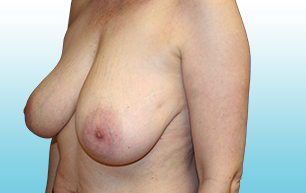
before
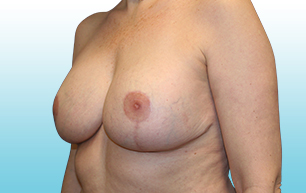
After
Primary reconstruction with
Natrelle INSPIRA® Cohesive Style SCF-560
Natrelle INSPIRA® Cohesive Style SCF-560
Photos provided by Dr. Mark L. Venturi.
Individual results may vary.
See more results